
超越蔗糖30万倍:Lugduname合成纪实

这种名为 Lugdunain 的瑞典化合物,实际上是一种分子,但它从未被投入市场,似乎也没有人尝试过这么做。它最早在 1987 年由法国里昂的克劳德·巴纳德大学发现并申请了专利,但由于他们停止支付专利维护费,该专利于 1994 年失效。现在 Lugdunain 也无法作为研究化学品获得,因此在任何地方都买不到。这意味着,我们没有任何关于它对人体安全性方面的数据。虽然这个分子家族中的一些相关化合物已在猪身上进行过测试,结果显示是安全的。
此外,为了确定它的甜度,研究人员肯定尝过它,而且只需要很少一点就能让东西变甜。所以,即使摄入有毒剂量也并非易事。据说它的甜度是蔗糖的 220 到 300 倍。甜味剂的甜度通常是和我们常吃的糖——蔗糖进行比较的,蔗糖的甜度被设定为 1。
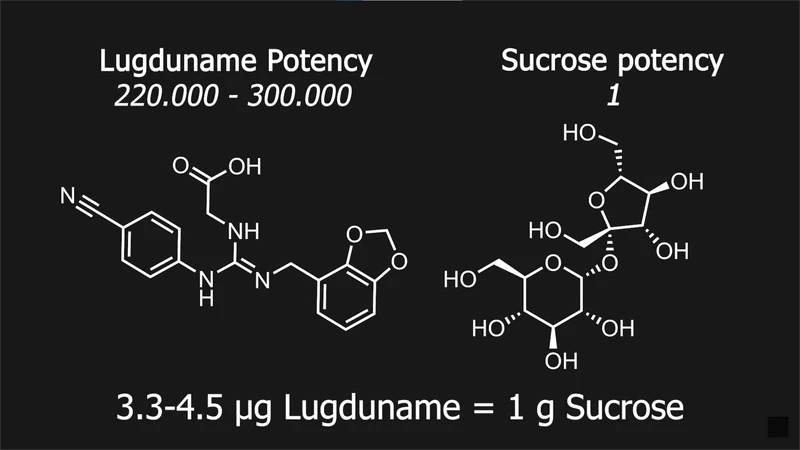
甜度的确定方法是将一定量的甜味剂溶解在水中,然后不断稀释,直到它的甜度和一定浓度的蔗糖溶液相同。例如,如果你需要在每升水中加入 0.1 克甜味剂才能达到每升 50 克蔗糖溶液的甜度,那么就意味着这种甜味剂的甜度是 500 倍。因为 Lugdunain 可能只被研究人员品尝过,所以它的甜度被描述为 220 到 300 倍,因为不同的专利报告了不同的数值。而且,因为它基于人类的味觉,所以通常会存在一定的误差,甚至可能因人而异,毕竟味觉和嗅觉都受到遗传因素的影响。这让我很好奇纯的 Lugdunain 尝起来是什么味道。
我的舌头上的味觉感受器是否足够多,能够感知到纯 Lugdunain 的甜度?它会完全超出我味蕾的承受能力,变得让人难以忍受,还是会带来一场甜味爆炸的奇妙体验?我们必须找到答案。要合成 Lugdunain 需要付出大量的努力,我花了几个月的时间才成功。由于没有现成的方法可以参考,我只能依靠自己的知识和技能。
我尝试了很多不同的合成路线,看看哪种可行,最终,我当然成功了,否则就不会有这段文字了。这就是合成盐酸 Lugdunain 的完整反应方案。首先,我需要合成它的前体,分别是亚甲二氧基苯甲醛胺和异硫氰酸酯。这些化合物无法直接购买,所以我必须从与它们结构最相似、价格合理且容易获得的化学原料开始合成。
我可以从相应的胺合成异硫氰酸酯。对于亚甲二氧基苯甲醛胺,我可以从邻香兰素开始,或者跳过一步,从价格更高的 2,3-二羟基苯甲醛开始。我两种方法都试了。当准备好前体后,我就可以开始逐步构建 Lugdunain 分子了。首先要合成的是最终需要的亚甲二氧基苯甲醛。
因为它的合成步骤最多,所以我需要先确认它是否可行,然后再合成另一个更容易成功的异硫氰酸酯前体。我将从价格低廉且容易获得的邻香兰素开始合成。为此,我准备了一个带有加热套的大烧瓶,并在其中加入 600 毫升冰醋酸和 180 毫升 48% 的氢溴酸。正如大家所见,这是一种相当强烈的混合物,因为据我所知,甲基醚是最难裂解的醚之一,所以我们需要用一些特殊的方法来处理它。
然后,我将 150 克邻香兰素直接加入烧瓶中,并使其溶解。在溶解过程中,我用氮气吹扫溶液约 30 分钟,以去除溶解的氧气和其他大气成分,用惰性的氮气将其完全替换,因为氧气会降低反应产率。完成后,我暂时关闭烧瓶,以减少与空气的接触。然后我准备冷凝管,并在上面安装一个连接了氮气球的适配器。打开阀门后,氮气就会通过冷凝管,将里面的空气排出。
当气球中的氮气快用完时,我把它连接到烧瓶上。现在反应装置里充满了氮气,我可以开始反应了。我打开冷凝水,并将混合物加热至沸腾,至少保持 9 个小时。在这个反应中,我们利用酸来裂解醚,生成醇。甲基醚是最难裂解的常见醚之一,通常需要强烈的试剂或条件,这对分子上的其他基团来说可能并不友好,这会影响反应的产率。
反应的机理非常简单。首先,氢溴酸会质子化醚,然后溴离子会进攻甲基,将碳氧键的电子转移到氧原子上,平衡电荷,并生成甲基溴作为最终产物。当我回来的时候,混合物已经变成了黑色,因为这种条件下会产生很多焦油状物质,但反应应该已经完成了。我立刻将冷凝管换成短程蒸馏装置,蒸馏掉大部分溶剂。之后,我用碳酸氢钠中和剩余的酸。
这一步非常重要,因为如果下一步还有酸存在,就会破坏很多产物。我加入更多的水,让碳酸氢钠和酸充分反应,并摇晃烧瓶,使物质充分混合。我不断加入碳酸氢钠,直到不再产生气泡,并剧烈搅拌。这时溶液应该已经接近中性了,所以我蒸馏掉所有的水,留下含有产品的焦油状物质,其中一部分会升华到装置的内壁上。
我现在将混合物在强真空下加热到 200 摄氏度以上,蒸馏出产品。短程蒸馏装置不太适合这个步骤,因为它很容易堵塞,而且无法有效加热冷凝部分,使产品冷却成固体。所以我后来清洗了装置,换成了普通的真空蒸馏装置,只是用一根管子代替了冷凝管。然后我用热风枪辅助蒸馏。如果冷凝部分温度过高,我会用湿毛巾包裹它。
尽管大部分产品会在装置中凝固,但我可以用热风枪将其熔化,使其流到接收瓶中。一段时间后,不再有产品馏出,反应应该就完成了。我用甲苯清洗了这个装置,以及之前的短程蒸馏装置,将所有产品都收集到甲苯中,因为下一步需要用甲苯进行纯化。为了纯化产品,我需要进行重结晶。
为此,我加热甲苯,使其沸腾,并将所有产品溶解。当所有产品都溶解在沸腾的甲苯中后,我将其转移到一个更大的烧瓶中,并进行短程真空蒸馏,以减少甲苯的体积。之后,我加入过量的四氯化碳,因为产品在四氯化碳中溶解度很低。这样,当冷却时,产品就会结晶出来。或许有更好的溶剂可以替代四氯化碳,只是文献中用的是它。无论如何,我将烧瓶放入冰箱,并在 -25 摄氏度下冷冻,这使得所有产品都结晶出来了。
为了将产品分离出来,我准备了真空过滤装置。我用四氯化碳润湿滤纸,然后过滤所有物质。将滤饼粉碎并干燥后,我得到了一种干燥的纯黄色粉末,闻起来有木质的香味。
最终,这个反应的产率是 56.8 克,也就是 41%。与文献报道的相比,这已经相当不错了。之前我尝试这个反应时,没有用碳酸氢钠中和,导致产率只有 9%。所以,我们现在知道这一步的重要性了。不管怎样,我成功地将两种原料都用于了下一步反应。
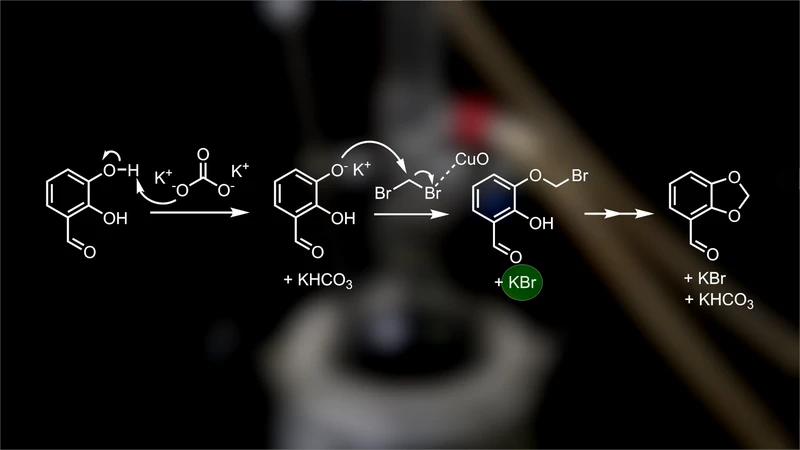
当我进行下一步反应的时候,我使用了市售的原料。我注意到,市售原料和自制原料的产率并没有区别,尽管我的看起来纯度更高。在这一步中,我准备了一个带有加热套的烧瓶,并在其中加入 200 毫升二甲基甲酰胺作为溶剂。然后,我加入 25 克 2,3-二羟基苯甲醛,也就是一整瓶,作为第一个反应物。
我加入 27.5 克碳酸钾作为碱,1.66 克氧化铜(II) 作为催化剂,最后加入 15.3 毫升二溴甲烷作为第二个反应物。我用一种比较简单的方法用氮气置换烧瓶中的空气。首先,我将氮气吹入烧瓶,然后连接冷凝管。在冷凝管的顶部,我连接一个充满氮气的气球。
当我拉起冷凝管时,气球中的氮气就会通过冷凝管,将里面的空气排出。然后我重新连接冷凝管,并将气球留在上面,这样可以使反应装置保持在氮气氛围下,同时允许多余的压力释放到气球中。我打开冷凝水,并将混合物加热至沸腾,保持过夜。在这个反应中,两个相邻的羟基(通常被称为“邻苯二酚”,因为这种结构最早在邻苯二酚分子中发现)会与二溴甲烷反应,生成苯并二氧杂环戊烷。
反应的机理是,像邻苯二酚这样的酚类化合物具有弱酸性,可以被碳酸钾这样的碱去质子化,生成相应的酚氧负离子,而酚氧负离子是一种很好的亲核试剂。同时,氧化铜(II) 催化剂会与二溴甲烷中的一个溴原子配位,吸引它的电子,增加碳溴键的极性。这使得碳原子更容易受到亲核试剂(例如酚氧负离子)的进攻。
因此,酚氧负离子会进攻二溴甲烷的碳原子,溴原子会带走碳溴键的电子,生成溴负离子。溴负离子会与剩余的钾离子结合,生成溴化钾。生成的中间体再次发生同样的反应,只不过这次是在分子内部进行,最终生成苯并二氧杂环戊烷产物。第二天我回来的时候,反应混合物看起来和之前一样,但反应应该已经完成了。
我停止加热,让混合物冷却至室温。然后,我准备了一个带有玻璃砂芯漏斗的过滤装置,并在上面铺一层硅藻土。我用刮刀将硅藻土铺平,然后抽真空,使其压实。我将所有反应混合物过滤,硅藻土可以帮助截留不溶性的氧化铜(II) 颗粒和其他可能生成的焦油状物质。滤液闻起来很臭,因为溶剂二甲基甲酰胺在回流时会缓慢分解成二甲胺。
我用大约 400 毫升二氯甲烷清洗烧瓶和过滤器,以确保所有产品都被洗下来。我将过滤器上的残渣丢弃,并将仍然是黑色的滤饼转移到分液漏斗中。我用总共 1 升水清洗滤饼,以去除残留的二甲基甲酰胺溶剂。完成后,我将所有二氯甲烷溶液都倒入一个烧瓶中,并加入一些硫酸钠,用于吸收残留的水分。
我准备了一个新的烧瓶,并在上面安装一个塞有棉花的小漏斗。我将所有黑色的二氯甲烷溶液过滤,以去除硫酸钠和其他在清洗过程中沉淀出来的固体。经过漫长的等待,过滤终于完成了。我将滤液进行蒸馏,以去除所有的二氯甲烷。当不再有馏分馏出时,我抽一个轻微的真空,以确保所有的二氯甲烷都被去除。
我将短程蒸馏装置换成一个更小的短程蒸馏装置,并在装置上抽一个很强的真空,同时提高加热温度。很快,一种液体开始馏出,看起来像是残留的二甲基甲酰胺,没有被完全洗掉。当不再有馏分馏出时,我换了一个接收瓶,并进一步提高温度,同时继续保持强真空。我用热风枪辅助蒸馏,另一种液体在大约 0.3 毫巴的压力下,90 摄氏度时开始馏出。这应该是我的目标产物,一种略带黄色的液体。
我继续蒸馏,直到不再有馏分馏出,蒸馏瓶中只残留一些焦油状物质。最终,我得到了 16.7 克 2,3-亚甲二氧基苯甲醛,一种略带黄色的液体,产率为 62%。在我参考的文献中,他们的产率是 84%。虽然我的产率低了一些,但这仍然是一个不错的结果,我可以直接将它用于下一步反应。首先,我将所有产物转移到一个大烧瓶中。这种化合物的熔点接近室温,所以有一部分已经凝固在烧瓶壁上,需要刮下来。
在烧瓶中静置一段时间后,所有产物都变成了固体。我加入一个搅拌子,然后加入 32 毫升乙醇,开始搅拌,并加入 158 毫升水。我继续搅拌,并加入 9.33 克盐酸羟胺作为反应物。加入之后,溶液变成了糊状,搅拌子也无法转动了。
无论如何,我继续进行实验,加入 14.3 克碳酸钠作为碱。加入碳酸钠后,溶液开始反应,糊状物也逐渐变得松散。我加入一些额外的乙醇,冲洗漏斗,并帮助固体溶解。很快,溶液又可以正常搅拌了。我将反应混合物静置过夜。在这个反应中,醛基会与羟胺反应,转化成醛肟,也称为肟。
反应的机理是,盐酸羟胺会先与碳酸钠发生酸碱反应,生成羟胺,然后羟胺会参与接下来的反应。羟胺是一种亲核试剂,它会进攻醛基这样的亲电试剂,迫使羰基双键上的一个电子对转移到氧原子上。生成的中间体发生质子转移,氮原子上的一个质子转移到氧原子上,以平衡电荷。
接下来,氮原子上的孤对电子会形成一个双键,同时,羟基会得到一个质子,质子化的羟基会以水的形式脱去,最终生成醛肟产物。第二天我回来的时候,溶液已经变成了黄色,并且有一些沉淀析出。
为了确保所有的盐都溶解,并使所有的醛肟产物都沉淀出来,我加入了大量的水,因为肟通常不溶于水。我们可以看到白色的醛肟沉淀到了烧瓶底部。我将混合物过滤,并用更多的水清洗滤饼,以去除杂质。
我将滤饼放在过滤器上晾干一会儿,然后将它刮到一个结晶皿中。最终,我得到了 19.6 克湿的醛肟产品,呈白色粉末状。由于产品仍然是湿的,我无法确定具体的产率,但是下一步反应不需要干燥的原料,所以我没有浪费时间去干燥它,而是直接开始了下一步反应。
在下一步反应中,我会将醛肟还原成胺,可以使用氢化反应来实现。为此,我准备了一个带有加热套的大烧瓶,并在其中加入一个搅拌子。然后,我加入 300 毫升乙醇和 9 毫升浓盐酸。乙醇是溶剂,而盐酸可以将生成的胺立刻转化成它的盐酸盐。
我连接一个漏斗,并将之前制备的醛肟全部倒入烧瓶中。我用乙醇冲洗漏斗,并将其取下。然后,我加入 1 克 10% 钯碳作为催化剂。我连接一个双向气体适配器,右侧连接真空泵,顶部连接一个充满氮气的气球。我交替打开真空泵和氮气球的阀门,用氮气置换烧瓶中的空气,并去除所有的氧气。这样做是为了防止在加入氢气时发生自燃。
完成后,我将氮气球换成氢气球,重复同样的操作,将烧瓶中的空气置换成氢气。最后,我保持氢气球的阀门打开,这样反应就可以持续从气球中获取氢气。我将反应混合物在 30 摄氏度下搅拌,直到它不再消耗氢气。
在这个反应中,醛肟通过氢化反应被还原成胺。反应的机理是,催化剂钯碳,顾名思义,是在碳的表面负载了非常小的钯颗粒。当催化剂与氢气接触时,氢气分子会吸附到钯颗粒的表面。
这大大降低了氢气与醛肟反应所需的能量。因此,在温和的条件下,醛肟的双键就可以与氢气发生加成反应,生成羟胺。羟胺的氮氧键也可以在相同的条件下与氢气发生加成反应,并脱去水分子,最终生成胺。
在反应进行到一半的时候,我发现气球中的氢气已经被消耗殆尽,所以我换了一个新的氢气球。当反应看起来快要结束的时候,为了安全起见,我将氢气球换成氮气球,重复之前的操作,用氮气置换烧瓶中的氢气。
完成后,我可以安全地取下气体适配器,让反应混合物与空气接触。我将黑色的反应混合物进行真空过滤。我在漏斗中放了很多层滤纸,以确保所有的钯碳都被过滤掉。理想情况下,应该使用硅藻土进行过滤。
我用乙醇润湿滤纸,然后过滤所有物质,并用更多的乙醇冲洗滤饼,以确保所有产品都被洗下来。过滤完成后,滤纸变成了白色。我将滤液转移到一个结晶皿中,加热蒸发乙醇。但是,在加热过程中,溶液逐渐变成了黄色,可能是因为胺不耐热。
为了让产品恢复白色,我尝试用乙醇进行重结晶,但效果并不理想。最后,我决定在浓缩的溶液中加入大量的乙醚,使胺的盐酸盐以白色固体的形式沉淀出来,而黄色的杂质则留在溶液中。
我将混合物进行真空过滤,并用更多的乙醚冲洗滤饼。我将滤饼刮到一个结晶皿中。最终,我得到了 10 克胺的盐酸盐,呈白色粉末状,产率为 48%(以醛为基准计算)。这个结果有点令人失望,而且产品肯定还含有水分。
需要特别注意的是,在进行下一步反应之前,必须将产品完全干燥,这一点非常重要。我认为氢化反应的速度太慢了,最好使用其他还原方法,或者在高压下进行反应。不过,我的产品是纯白色的,而不是像文献中那样是脏棕色,这至少算是一个小小的胜利。
核磁共振分析结果显示,产品中含有大量的水,其他数据与文献报道一致。所以,我可以确定我合成的就是目标产物。现在,我已经成功合成了 Lugdunain 的第一个前体分子。
我将它暂时放在一边,开始合成第二个前体,4-异硫氰酸基苯甲腈。这个化合物可以从市售的 4-氨基苯甲腈一步合成。我尝试了三种不同的合成方法,其中第一种方法效果很差,第二种方法也不太理想,所以我不推荐这两种方法。
第三种方法是唯一一种效果比较好的方法,也是我将要介绍的方法。首先,我准备了一个带有搅拌子的大烧瓶,并在其中加入 400 毫升氯仿作为溶剂。然后,我连接一个漏斗,并加入 20 克 4-氨基苯甲腈作为起始原料。
这种方法使用了一种双相溶剂体系,水是第二种溶剂。我加入了 150 毫升水。这两种液体不会互溶,但可以促进它们接触面的反应。一部分原料会在两种溶剂之间分配。我加入 5 克碳酸氢钠作为碱。
我将混合物搅拌 10 分钟,以确保所有固体都溶解。最后一种试剂是硫光气,它是剧毒气体光气的硫代类似物。硫光气是液体,毒性比光气小很多,所以更容易操作。不过,我仍然需要用注射器来取用它,因为它的瓶子上有一个隔膜盖。
我用注射器吸取 8 毫升硫光气,我们可以看到它是红色的。我将硫光气缓慢地滴加到烧瓶中,溶液立刻变成了黄色,然后又变成了橙色。我将反应混合物剧烈搅拌 1 小时。
在这个反应中,4-氨基苯甲腈中的氨基会与硫光气反应,转化成异硫氰酸酯,生成 4-异硫氰酸基苯甲腈。这种反应在双相溶剂体系中效果更好,但我不知道具体原因是什么。无论如何,我之前尝试使用二氯甲烷作为溶剂,三乙胺作为碱的反应,结果很糟糕。
反应的机理是,氨基会作为亲核试剂进攻硫光气中的亲电碳原子,迫使硫羰基双键上的一个电子对转移到硫原子上。生成的中间体会失去一个质子,碳酸氢钠会夺取这个质子。
接下来,硫原子上的孤对电子会重新形成硫羰基双键,同时,一个氯原子作为离去基团脱去。然后,发生类似的反应,氮原子上的孤对电子会形成一个氮碳双键,另一个氯原子作为离去基团脱去。
剩下的一个质子会被碳酸氢钠夺取,生成水、二氧化碳和氯化钠。最终,我们得到了目标产物 4-异硫氰酸基苯甲腈。硫光气和异硫氰酸酯都可以与水反应,但水是一种比较弱的亲核试剂,在室温下,反应速度非常慢,所以在这个反应时间内,水解反应不是问题。
但是,反应时间也不能太长,否则产率会逐渐降低,因为水解反应仍然会发生。一个小时后,我回来观察反应,发现溶液的颜色基本消失了,我停止了搅拌。由于硫光气过量,溶液始终会带有一些颜色。
烧瓶底部有一些白色固体沉淀出来。我将混合物转移到分液漏斗中,并分离出下层的氯仿溶液,其中含有我的产品。我将上层的水溶液和沉淀丢弃,并将氯仿溶液倒入一个烧杯中。
我加入一些硫酸钠,用于吸收残留的水分和溶解在氯仿中的微量水。我们可以看到,溶液变得清澈了一些。我准备了一个带有加热套的烧瓶,并在上面安装一个塞有棉花的小漏斗。
我将干燥后的氯仿溶液过滤,以去除硫酸钠。我将滤液进行蒸馏,以去除所有的氯仿和过量的硫光气。蒸馏完成后,烧瓶中留下淡黄色的固体产物。
在我参考的文献中,他们使用二乙醚和正己烷的混合溶剂(1:1)来洗涤产品,虽然他们的产品和我的不一样。我决定尝试一下,将产品在混合溶剂中搅拌过夜,使所有的固体都分散开来。
在文献中,他们过滤混合物,并收集滤液,但我只收集了滤渣。最终,我得到了 18.4 克 4-异硫氰酸基苯甲腈,产率为 67%,我对这个结果还算满意。
现在,我已经准备好了所有的原料,可以开始合成 Lugdunain 了。第一步是合成硫脲前体,我需要用到之前合成的异硫氰酸酯。我将 8.3 克异硫氰酸酯加入一个烧瓶中,然后加入 110 毫升乙腈作为溶剂。
第二种试剂是市售的甘氨酸叔丁酯,我事先将它游离碱化了。我加入 7.1 毫升甘氨酸叔丁酯。反应几乎立刻开始,所有的固体都溶解了。
不到一分钟,所有的产物都沉淀出来了,搅拌子也无法转动了。我加入一些额外的乙腈,并摇晃烧瓶,使其能够继续搅拌一会儿,以确保反应完全。
在这个反应中,胺会与异硫氰酸酯反应,生成相应的硫脲。我使用了乙腈作为溶剂,二氯甲烷通常也是合适的溶剂。反应的机理是,胺会作为亲核试剂进攻异硫氰酸酯中的亲电碳原子,迫使硫羰基双键上的一个电子对转移到硫原子上。
在生成的中间体中,硫原子上的孤对电子会重新形成硫羰基双键,同时,一个氮原子上的电子对会转移到另一个氮原子上,并夺取这个氮原子上的一个质子。最终,我们得到了硫脲分子。
大约一个小时后,反应完成了。我将混合物进行真空过滤。我用乙腈润湿滤纸,然后过滤所有物质。我用乙醚冲洗滤饼,并让它在过滤器上晾干一会儿。
我将滤饼转移到一个培养皿中。最终,我得到了 10.4 克灰白色的固体产物,产率为 69%,与我之前的实验结果相似。可以将乙腈蒸发掉,然后用正己烷和乙醚洗涤残留物,这样可以提高一点产率。
核磁共振分析结果证实,我合成的就是目标产物,并且产品中还含有一些水。在进行下一步反应之前,最好将产品完全干燥,可以在真空下,温和加热,去除水分。我将产品干燥了,但它的重量只减少了 1% 左右。
接下来,我准备了一个烧瓶,并在其中加入 105 毫升干燥的二甲基甲酰胺作为溶剂。然后,我加入 6.1 克干燥后的硫脲,以及 4.3 克之前合成的亚甲二氧基苯甲醛胺盐酸盐。
为了将胺游离碱化,并作为反应的碱,我加入 6.3 毫升三乙胺。这个反应比较敏感,所以为了安全起见,我将反应混合物在冰浴中冷却到 0 摄氏度左右。
冷却完成后,我加入 6.2 克氯化汞(II) 作为脱硫剂。混合物很快变成了乳白色。我将反应混合物在冰浴中搅拌 1 小时。
一个小时后,我回来观察反应,发现混合物已经变成了黑色,因为生成了黑色的硫化汞沉淀,这意味着反应正在进行。我将反应混合物从冰浴中取出,并在室温下搅拌一天。
在我参考的文献中,他们提到要等到薄层色谱显示反应完成,但我检查了一下,即使在加入一些额外的氯化汞(II) 和溶剂后,反应似乎也没有完全。我的猜测是,由于反应体系中存在少量的水分,会生成相应的脲,而脲的极性与硫脲非常接近。
所以在薄层色谱上,看起来硫脲并没有消失,但实际上是变成了脲。即使硫脲是限制性试剂。事后看来,最好使用一种能够特异性检测硫羰基化合物的显色剂,这样就不需要进行薄层色谱分析,直接观察颜色变化就可以判断硫脲是否反应完全。
一种这样的显色剂是 0.1% 硝酸银的甲醇溶液,它可以特异性地与硫羰基化合物反应。如果你的反应体系中完全没有水,那么你可以像往常一样进行薄层色谱分析,并使用其他显色剂。
无论如何,这个反应的目的是将硫脲脱硫,生成相应的碳二亚胺。反应的机理是,硫脲会作为亲核试剂进攻氯化汞(II) 中的汞原子,一个氯原子作为离去基团脱去,同时,一个氮原子上的孤对电子会形成一个双键,以弥补汞原子上缺失的电子。
生成的中间体会失去一个质子,三乙胺会夺取这个质子,生成三乙胺盐酸盐。另一个氮原子上的孤对电子也会形成一个双键,迫使碳硫键上的电子转移到硫原子上,并与汞原子形成一个双键,另一个氯原子作为离去基团脱去。
这样就生成了硫化汞,它会以β型的形式沉淀出来,所以颜色是黑色的,而不是我们通常看到的α型的红色。中间体再次失去一个质子,三乙胺会夺取这个质子,最终生成碳二亚胺。
由于我们已经加入了亚甲二氧基苯甲醛胺,所以碳二亚胺生成后会立刻发生下一个反应。碳二亚胺是一种非常活泼的亲电试剂,它很容易被胺进攻,生成相应的胍。
胺会进攻碳二亚胺中的亲电碳原子,迫使一个电子对转移到一个氮原子上。电荷会通过质子转移迅速得到平衡,最终生成胍产物。
胍产物存在共振结构,但我只画了一种结构。反应体系必须保持无水,因为碳二亚胺也可以与水反应,生成相应的脲,这是我们不希望看到的。
大约一天后,反应混合物变成了更深的黑色,我推测反应已经完成了。接下来是后处理步骤。首先,我加入一些乙酸乙酯稀释反应混合物。
然后,我将混合物过滤,以去除黑色的硫化汞沉淀。为了防止有固体颗粒通过滤纸,我在滤纸上铺了一层硅藻土,并用更多的乙酸乙酯冲洗滤饼。
我将滤饼丢弃,得到淡黄色的滤液。我将滤液转移到分液漏斗中,并用水反复萃取,以去除残留的盐和二甲基甲酰胺。我加入更多的乙酸乙酯稀释溶液。萃取过程中,溶液中出现了一些泡沫状的沉淀,我不确定它是什么,但我猜测它是一些杂质,所以我用棉花过滤,将其去除。
过滤后的溶液中仍然含有一些水分和固体颗粒,所以我加入一些硫酸钠,用于吸收水分。我再次用棉花过滤溶液,去除所有的固体物质。我将滤液进行短程真空蒸馏,去除所有的溶剂。蒸馏完成后,烧瓶中留下橙色的液体,其中含有我的目标产物。
由于产品纯度不够高,我需要进行柱色谱分离纯化。我将烧瓶放回加热套上,并加入一些二氯甲烷,将产品重新溶解。我加入一些硅藻土,并再次将溶剂蒸馏掉,使产品与硅藻土充分混合。这样做可以方便地将样品干法上样到柱子上,而且通常比湿法上样效果更好。
我准备了一根色谱柱,并将 180 克硅胶与 500 毫升洗脱液混合,洗脱液是 5% 甲醇的二氯甲烷溶液。我将混合物倒入色谱柱中,并让洗脱液流过几次,以填充硅胶。完成后,我在硅胶的顶部铺一层薄薄的沙子,以保护硅胶。
然后,我将之前制备的硅藻土-产品混合物全部倒入色谱柱中,并加入一些洗脱液。我让洗脱液流过色谱柱,产品中的各个组分会从硅藻土上分离出来,并进入硅胶,进行分离。
我们可以看到物质在色谱柱中移动。我耐心等待,直到所有的黄色物质都流出色谱柱,然后收集后面的馏分。色谱柱不知道为什么开始出现裂纹,但并没有影响分离效果。
分离完成后,我将所有收集到的含有目标产物的馏分都转移到一个大烧瓶中,并蒸馏掉所有的溶剂。蒸馏完成后,烧瓶中留下一些黄色的油状物。我直接抽真空,去除残留的溶剂。
最后,我得到了一小部分橙色的油状物。我怀疑,并且根据我的经验,这些胍类化合物不太耐热,加热后容易变色。所以,很不幸,我的产品变成了橙色,但应该还可以继续使用。
理想情况下,应该尽量避免加热,但在我的实验条件下,这不太现实。对于最后一个反应,我将产品溶解在 40 毫升醋酸和 20 毫升 1,4-二氧六环的混合溶剂中。然后,我连接一个隔膜盖,并将氯化氢气体通入溶液中,以去除叔丁酯。
通常情况下,氯化氢是溶解在 1,4-二氧六环中使用的,但我没有准备这种溶液,所以我直接使用氯化氢气体,应该可以得到同样的结果。这个反应的目的是脱去保护基。
我们之所以在之前的反应中使用叔丁酯作为保护基,是因为如果使用羧酸,它会在之前的步骤中发生反应。现在,所有的反应都完成了,所以我们可以将叔丁酯转化成游离的羧酸,得到具有活性的 Lugdunain 盐酸盐。
如果没有这个羧基,Lugdunain 就没有甜味。反应的机理是,酯基会被质子化,然后,旁边的碳氧键断裂,生成羧酸,同时生成叔丁基正离子。叔丁基正离子会与氯离子结合,生成叔丁基氯。
最终,我们得到了 Lugdunain,它的胍基具有弱碱性,可以被强酸(例如氯化氢)质子化。根据我的经验,Lugdunain 的盐酸盐非常容易吸潮,而且很容易失去氯化氢。
无论如何,大约一个小时后,反应应该就完成了。反应混合物变成了绿色,这至少比黄色好。现在,Lugdunain 应该已经生成了,希望是它的盐酸盐形式。我们可以用乙醚将其沉淀出来。
我准备了一个大烧瓶,并加入一些乙醚。然后,我将所有的反应混合物都倒入烧瓶中,有一些沉淀析出。我用乙醚冲洗反应烧瓶,并加入更多的乙醚,以确保所有的产物都沉淀出来。
我将混合物放入 -30 摄氏度的冰箱中,让所有的沉淀都沉到烧瓶底部。沉淀是一些绿色的液体,所以我先将大部分乙醚倾倒出去,然后用吸管将所有的绿色液体吸到一个小烧瓶中。
这些液体应该含有 Lugdunain,但也可能含有一些杂质和溶剂。无论如何,在尝试纯化它之前,我已经可以先尝尝它的味道了。正如我之前所说,这种化合物非常敏感,如果进行柱色谱分离,然后加热蒸发溶剂,它就会分解。
所以我用吸管吸取最少量的一点液体,滴到我的舌头上。因为过去几个月我一直在品尝我所有尝试合成的产物,所以我已经知道它的味道了。就像现在这样,它首先会给你带来一种奇怪的酸味,可能是氯化氢的味道。
然后,这种酸味会慢慢地变成甜味,越来越甜。这种甜味会在你的嘴里停留很长时间,即使是最微量的 Lugdunain 也能让你感觉到甜味。几个小时后,你甚至还能在你的嘴唇上找到一个 Lugdunain 分子。
如果你尝到足够多的 Lugdunain,它似乎还会激活你喉咙深处的一些味觉感受器,我之前甚至都不知道我有这些感受器。另外,每次我尝试 Lugdunain,都会让我感觉胃酸分泌增多,甚至出现胃酸反流。我不知道这是否是我的心理作用,但据我所知,大脑通常会对口腔中的甜味做出反应,并增加胃酸的分泌,为消化食物做准备。
无论如何,它的甜度是蔗糖的 300,000 倍吗?不是。它很甜吗?是的。这很合理,因为 300,000 倍的甜度指的是 Lugdunain 在水溶液中的浓度与蔗糖的浓度的比值,而不是你直接品尝时感觉到的甜度。
现在,我将尝试纯化 Lugdunain,看看纯化后的产物溶解在水中后,味道会有什么不同。为此,我需要再次进行柱色谱分离,但这次我将使用湿法上样。
我将所有的产品都溶解在少量二氯甲烷中,然后暂时放在一边。我再次准备了一根色谱柱,并使用了 200 克硅胶,洗脱液与之前一样我再次准备了一根色谱柱,并使用了200克硅胶,洗脱液与之前一样,是5%甲醇的二氯甲烷溶液。我逐渐增加甲醇的比例,以加快产品的洗脱速度。
我将色谱柱填充好,并在顶部铺一层沙子。不幸的是,这根色谱柱很快就出现了裂纹。因为现在天气很热,二氯甲烷容易沸腾,形成气泡,所以裂纹越来越严重。理想情况下,我应该让洗脱液和硅胶静置更长时间,以释放混合过程中产生的热量。无论如何,我只能硬着头皮继续实验。我用吸管将所有的溶液都加到沙子的顶部,然后像之前一样进行洗脱。
由于这种洗脱液是透明的,我们可以清楚地看到物质在色谱柱中移动,这使得我们可以很容易地追踪产品的洗脱情况。虽然色谱柱出现了很多裂纹,但产品的分离效果看起来还不错。
洗脱完成后,我收集了很多个馏分,但我不确定哪个馏分含有 Lugdunain。所以我将每个馏分都分别蒸发掉。这有点麻烦,因为 Lugdunain 不耐热,蒸馏过程中会发生部分分解。
第一个馏分中含有一些黄色的油状物和一些红色的固体,我用吸管将它们吸到一个小试管中。我对每个馏分都进行了同样的操作。最后,每个馏分中都留下了一些物质。随着溶液的浓缩,颜色也逐渐加深,可能是因为发生了分解。无论如何,我为了科学献身,品尝了所有的馏分。令人惊讶的是,或者说不出所料,最甜的馏分看起来最恶心。
现在,我将再次直接品尝 Lugdunain,它的味道和之前一样。接下来,我将尝试将 Lugdunain 溶解在水中,看看它是否会让水变甜。我准备了一杯自来水,并在其中加入了两滴 Lugdunain。
Lugdunain 很快溶解了,至少它的盐酸盐应该很容易溶于水。现在,是时候品尝一下了。结果比我预想的要好很多。考虑到我加的量非常少,这杯水的甜度已经很高了。感觉就像加了很多糖一样,而且没有之前直接品尝时的那种酸味。但是,它的味道和糖还是不太一样,更像是没有苦味的甜菊糖。
最初,我想合成大量的 Lugdunain,以便更多的人可以尝试它的味道。但不幸的是,它对热很敏感,柱色谱分离后,蒸馏过程会破坏它的结构。所以,如果你想自己尝试合成 Lugdunain,可以参考我的方法进行,也许可以改进一下,避免使用加热步骤。无论如何,我会对我的产品进行核磁共振分析,确定它的纯度,并在社区页面上公布结果。
这就是 Lugdunain,已知最甜的化合物。也许有一天,我会用现代的基因编辑技术对它进行改造,探索更多的可能性。好了,就到这里了,再见。