
农科院棉花所:东方粘虫染色体水平的基因组组装
编译:微科盟-茹梦,编辑:微科盟悄**、江舜尧。
微科盟原创微文,欢迎转发转载,来源《基因组Genome》公众号。
需要原文可私信获取!
导读
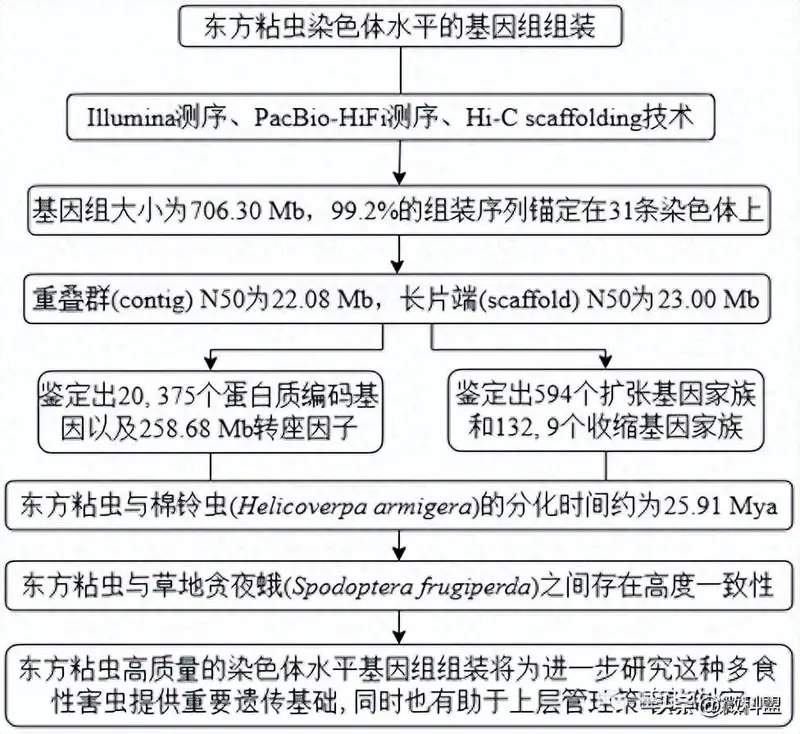
东方粘虫(Mythimna separata)是一种极具破坏性的多食性害虫,寄主范围广,严重威胁农业生产安全。在本研究中,使用Illumina、PacBio-HiFi长测序和Hi-C scaffolding技术组装了高质量的染色体水平基因组。基因组大小为706.30 Mb,重叠群contig N50为22.08 Mb,99.2%的组装序列锚定在31条染色体上。此外,还鉴定出20,375个蛋白质编码基因和258.68 Mb转座因子。东方粘虫染色体水平的基因组组装为未来进行该昆虫的研究提供了重要的遗传资源。
论文ID
原名:Chromosome level genome assembly of oriental armyworm Mythimna separata
译名:东方粘虫染色体水平的基因组组装
期刊:Scientific Data
IF:9.8
发表时间:2023年9月
通讯作者:姬继超,高雪珂,雒珺瑜,崔金杰
通讯作者单位:中国农业科学院棉花研究所
DOI: 10.1038/s41597-023-02506-3
实验设计
结果
东方粘虫Mythimna separata(鳞翅目,夜蛾科)是一种臭名昭著的多食性害虫,广泛分布在亚洲、澳大利亚、新西兰和几个太平洋岛屿(图1a)。这种害虫的寄主范围很广,对作物的生产构成严重威胁,尤其是水稻、玉米和小麦(图1b)。2012年至2013年,东方粘虫在中国爆发,威胁到17.437亿公顷农田,近年来这种威胁一直持续存在。在东方粘虫感染的其他国家和地区也出现了这种情况。近年来,随着全球气候、作物种植结构、品种分布和栽培体系的变化,东方粘虫在适应性、爆发性和危害性方面表现出了新的特点。由于其群居性、迁移能力强、多食性和暴食性,东方粘虫于2020年被中国农业农村部列入一级农作物病虫害名录。
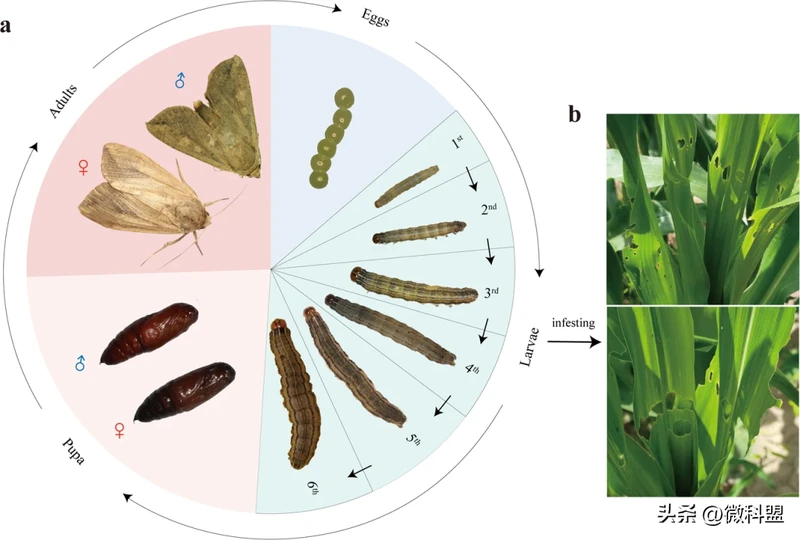
图1 东方粘虫的发育期及危害
先前的研究表明,多食性昆虫通过诱导解毒酶相关基因表达的变化,对不同寄主植物产生的有毒次级代谢产物做出反应。这种变化可能会增强多食性昆虫适应寄主植物的能力,并产生对杀虫剂的抗性。然而,由于基因组资源的稀缺,上述假说无法在东方粘虫中得到验证。尽管在2022年和2023年发表了几个东方粘虫基因组组装,但这些基因组组装的组装方法和质量存在显著差异。因此,一个高质量的染色体水平的基因组是提供遗传资源和深入研究东方粘虫解毒作用和宿主适应分子机制所必需的,这将有助于为优化东方粘虫的管理策略提供理论支持。
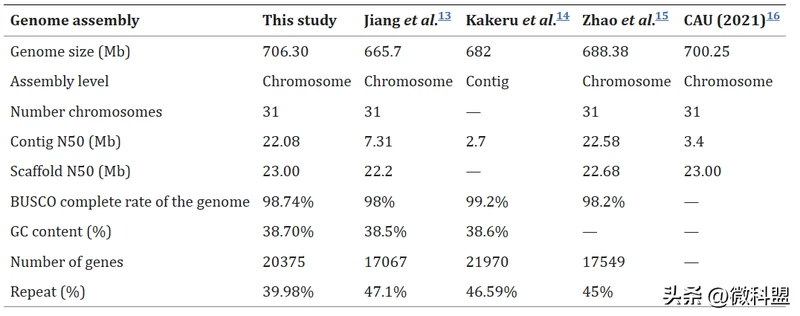
在本研究中,研究人员通过使用Illumina短读长、PacBio高保真(HiFi)读长和高通量染色体构象捕获(Hi-C)数据的组合,组装了东方粘虫的高质量染色体水平基因组(表1)。基因组组装由172个重叠群(contigs)组成,总长度为706.30 Mb,其中contig N50为22.08 Mb。此外,99.2%的初级组装(700.63 Mb)锚定在31条染色体上,长片端scaffold N50为23.00 Mb。研究人员还确定了258.68 Mb的串联重复序列,占基因组组装的36.63%。共获得20,375个蛋白质编码基因,其中98.53%进行了注释。系统发育分析结果表明,东方粘虫与棉铃虫(Helicoverpa armigera)的分化时间约为25.91 Mya。此外,东方粘虫基因组中还鉴定出594个扩张基因家族和1329个收缩基因家族。东方粘虫高质量的染色体水平基因组组装将为进一步研究这种多食性害虫提供遗传基础。
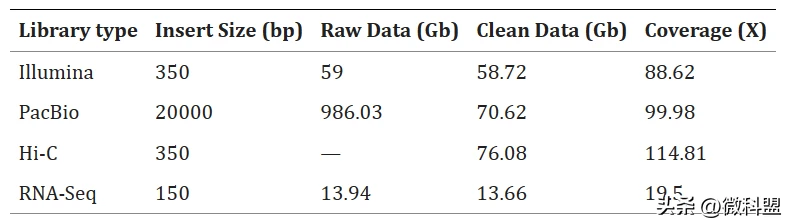
表1 东方粘虫基因组测序数据统计
方法
样本采集和基因组测序
东方粘虫是从中国河南省安阳市的玉米地里采集的,随后在26±1℃的恒温箱中饲养,相对湿度为70%,光周期为14小时照光及10小时黑暗(14hL:10hD)。使用基因组DNA小提试剂盒QIAamp DNA Mini Kit从单个表面灭菌的雄蛹中提取基因组DNA,用于Illumina和PacBio-HiFi测序,以防止其他个体和微生物的污染。从单个成年雄性个体中提取基因组DNA用于Hi-C测序。使用TRIzol试剂盒从成年个体中提取总RNA用于转录组测序。基因组DNA和RNA的纯度和完整性通过NanoDrop 2000C分光光度计和琼脂糖凝胶电泳(1.5%)进行验证。
在Illumina NovaSeq 6000平台上构建并测序具有350 bp插入片段的配对端文库。使用HTQC(v1.92.310)软件删除接头序列和低质量读取,之后总共获得58.72 Gb纯净读长(clean reads)用于后续分析。对于PacBio-HiFi测序,使用g-Tubes将基因组DNA剪切成约15 Kb的片段,并使用0.45×AMPure PB珠纯化,用于构建单分子实时测序文库(SMRT bell)。使用Sage ELF系统进行选择,以收集15–18 Kb的SMRT bell文库。在引物退火并将Sequel II DNA聚合酶与SMRT bell模板结合后,使用8 M SMRT小格在Sequel II系统上进行测序。 通过循环共识测序(CCS)模式获得986.03 Gb子读长并用于生成PacBio-HiFi读长。最后,共获得了70.62 Gb的CCS读长,平均读取长度为16.67 kb,东方粘虫基因组的覆盖率为99.98X。按照标准文库制备方案构建Hi-C文库,并在Illumina NovaSeq 6000平台上测序,获得了76.08 Gb的150 bp配对末端纯净读长。
基因组调查和组装
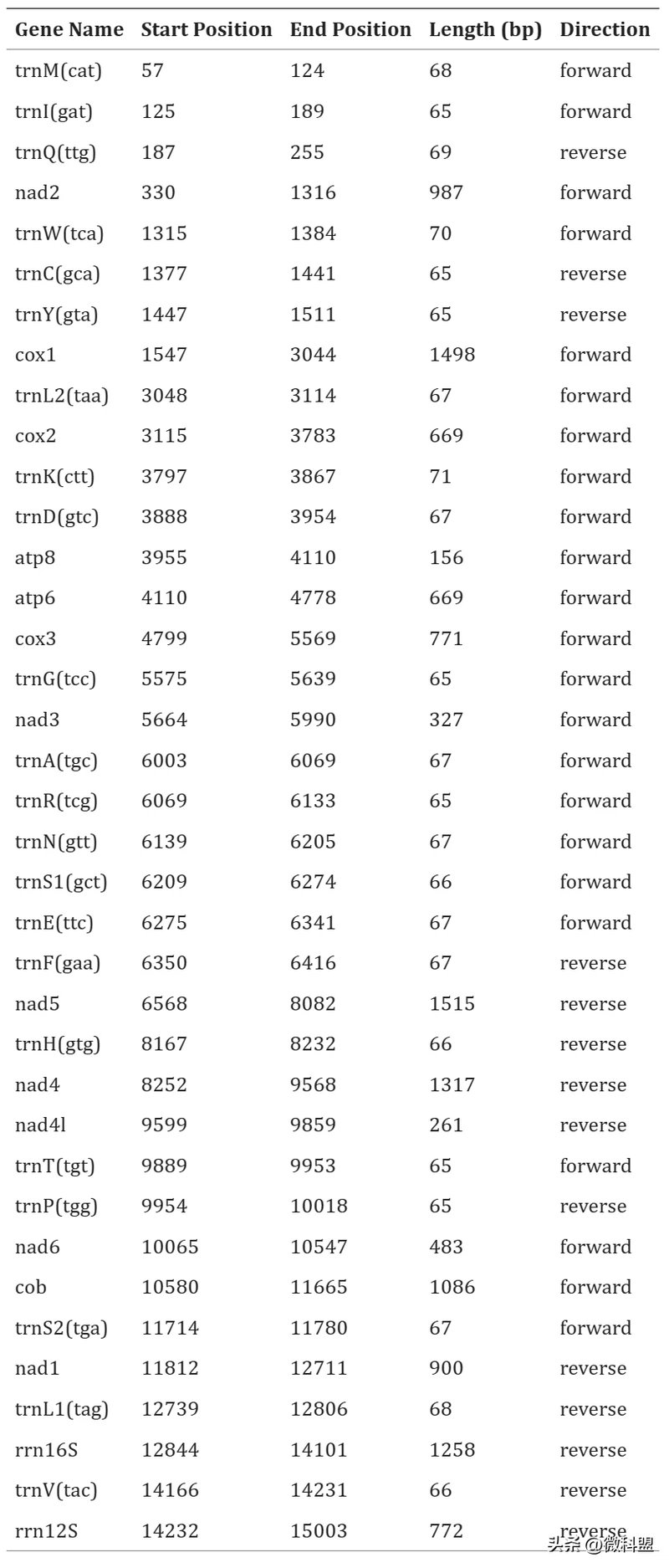
基因组调查对于估计主要特征至关重要,包括基因组大小、重复序列含量和杂合性。基于Illumina纯净短读长使用Jellyfish构建k-mer(k=19)频率,并使用GenomeScope进行基因组调查。东方粘虫的基因组大小估计为662.64 Mb,重复含量为39.00%,杂合度为0.76%(图2a)。随后,CCS读长被提交给Hifiasm,并使用默认参数进行组装。使用purge_dups过滤单倍型重复,参数为“−2 -T cutoffs -c PB.base.cov”,生成东方粘虫的基因组组装。该组装体由172个重叠群(contig)组成,总长度为706.30 Mb,contig N50为22.08 Mb 。使用具有默认参数的BWA(0.7.10)将纯净Hi-C读长与初级基因组组装进行比对。使用HiC Pro(v2.10.0)对唯一对齐的读长对进行进一步处理,以评估和消除无效读取对,包括悬空端、重连接、自循环和转储对。在默认参数下使用LACHESIS将88,824,108个用于长片端(scaffold)校正的有效相互作用对用于contigs聚类、排序和定向到染色体上。最后,147个scaffolds被锚定在31条染色体上,scaffold N50为23.00 Mb,占据700.63 Mb,占基因组初级组装的99.2%(图2b,c,表2)。此外,东方粘虫的线粒体基因组通过mitoZ和NOVO plasty软件进行组装,随后使用MITOS和GeSeq进行注释(图3a,表3)。
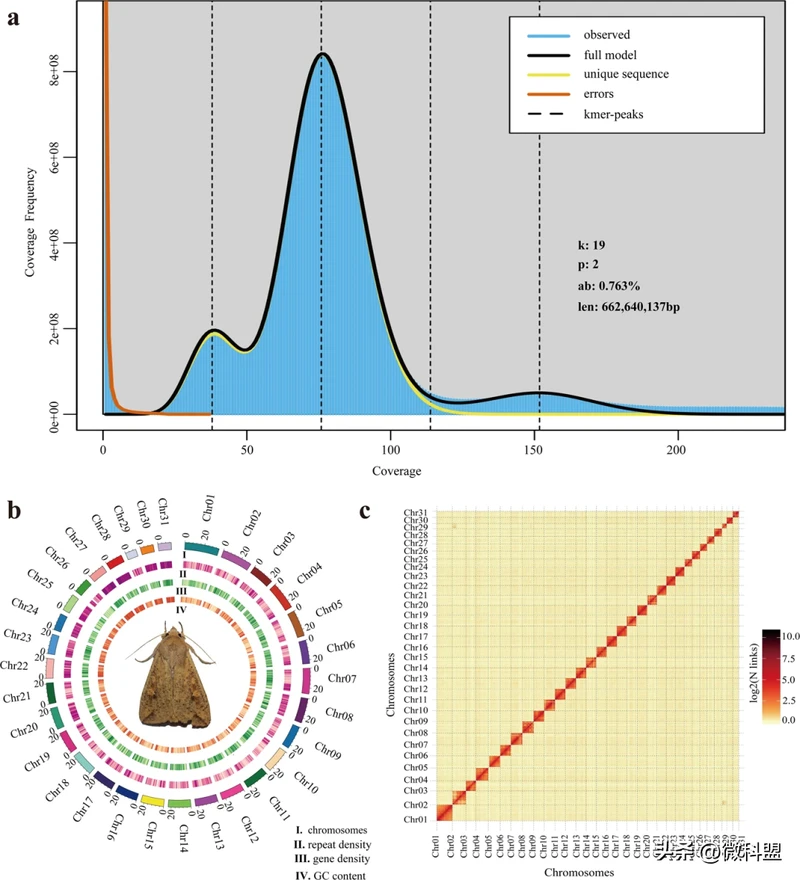
图2 东方粘虫基因组组装图谱。(a)19-mer分析的基因组范围图谱。(b)东方粘虫的基因组圈图。圈I表示染色体,圈II-IV表示每条染色体的重复密度、基因密度和GC含量。(c)东方粘虫的Hi-C交互热图。颜色表示相互作用信号的强度。颜色越深,强度越高。
表2 Hi-C组装结果统计
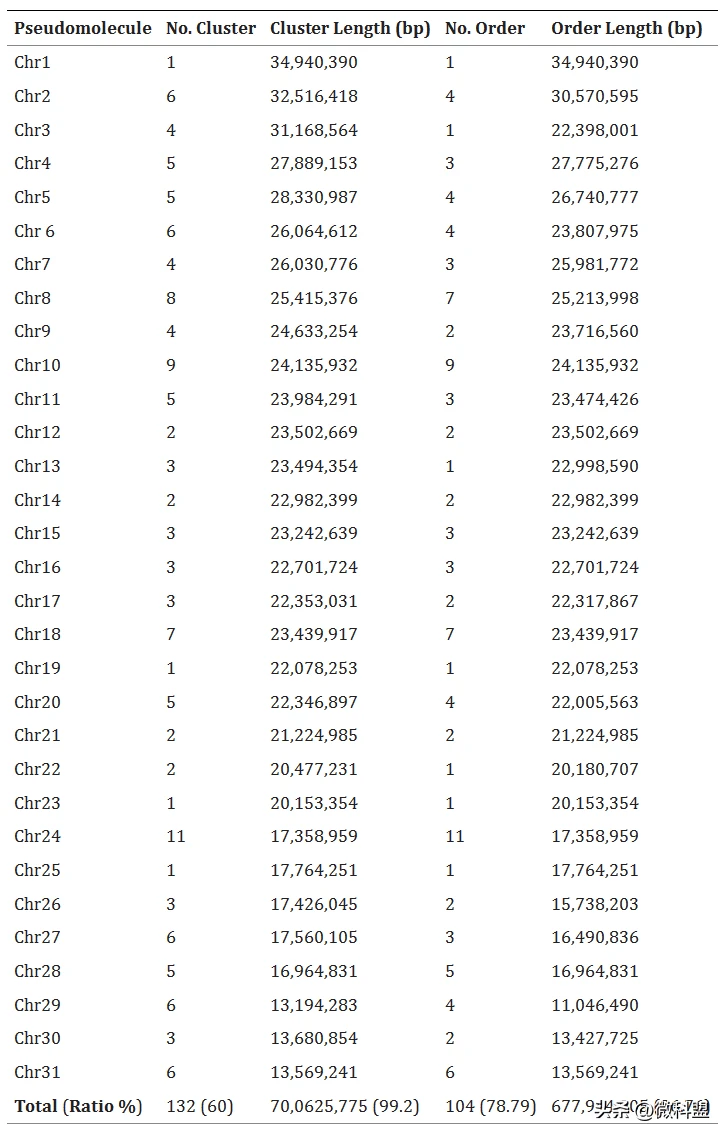
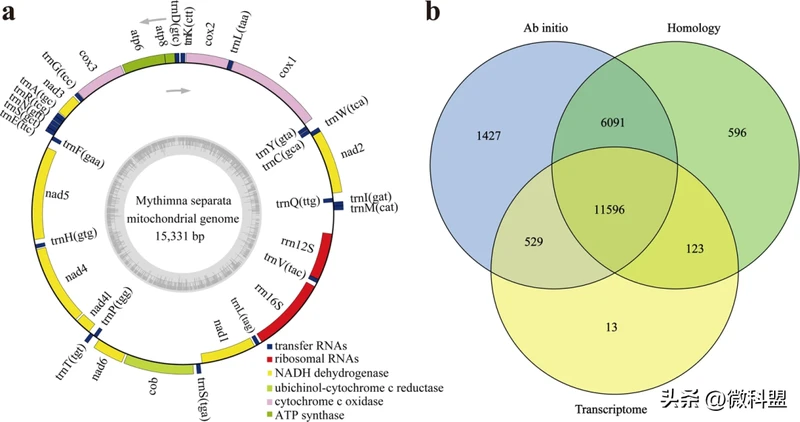
图3 东方粘虫线粒体基因组组装和蛋白质编码基因预测。(a)东方粘虫线粒体基因组圈图。基因图谱显示了37个不同功能群的注释基因。(b)三种预测方法得到的蛋白质编码基因的维恩图。
表3 东方粘虫线粒体基因组注释
基因组重复注释
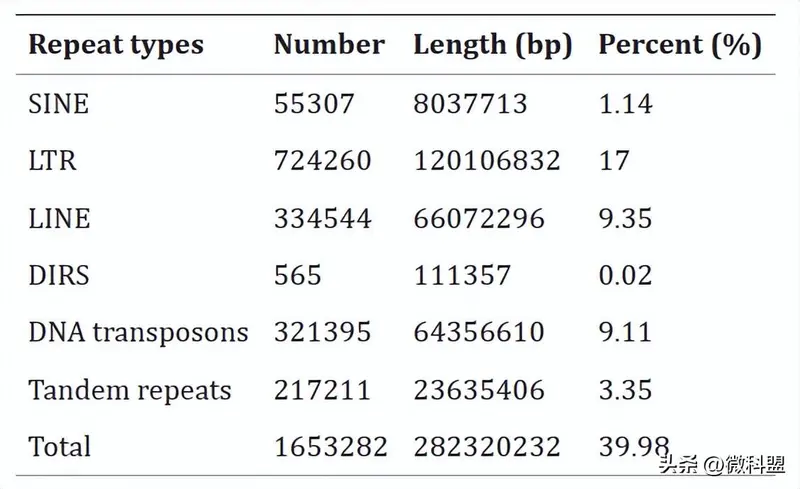
重复序列主要包括串联重复序列和散在重复序列,后者主要是转座因子(TE)。使用基于同源性和从头测序(de novo)方法对TE的重复序列进行注释。研究人员最初使用RepeatModeler和LTR_retriever在默认参数下为组装序列制定了一个从头测序重复库。随后使用PASTEClassifier对预测的重复序列进行分类,并将结果与Repbase、REXdb和Dfam的数据库相结合,构建去除冗余序列的物种特异性TE文库。通过使用RepeatMasker对文库进行同源性搜索以鉴定TE序列。总计获得了258.68 Mb的TE序列,占基因组组装的36.63%。此外,使用MISA和NCRF鉴定了23.64 Mb(3.35%)的串联重复序列(表4)。
表4 东方粘虫基因组重复元素统计
基因预测与功能注释
在消除东方粘虫基因组重复序列的干扰后,将三种方法(包括从头预测、基于同源性和基于转录组的方法)结合起来进行基因预测。使用Augustus和SNAP两个从头开始的基因预测软件工具,在默认参数下预测de novo基因模型。使用GeMoMa对从GenBank下载的鳞翅目昆虫的蛋白质序列进行基于同源性的基因预测,涉及家蚕(Bombyx mori)、棉铃虫(Helicoverpa armigera)、草地贪夜蛾(Spodoptera frugiperda)和斜纹夜蛾(Spodoptera litura)。对于基于转录组的基因预测,使用Trinity将RNA-seq读长组装成通用的基因数据库(unigenes),然后使用产生的unigenes通过PASA鉴定蛋白质编码基因。最后,在默认参数下使用EVidenceModeler将这三种方法获得的基因模型集成到一个统一的基因集中。最终,从东方粘虫基因组中鉴定出203,75个蛋白质编码基因(图3b)。
为了对蛋白质编码基因进行功能注释,研究人员使用阈值为1e−5的BLAST(v2.2.31)将预测基因与包括NR、GO、KEGG、EggNOG、KOG、TrEMBL、InterPro和Swiss-Prot在内的数据库进行比对。最后,98.53%(20075/20375)的蛋白质编码基因被注释(表5)。解毒相关基因细胞色素P450(P450)、ATP结合盒(ABC)、羧基/胆碱酯酶(CCE)、UDP糖基转移酶(UGTs)和谷胱甘肽-S-转移酶(GST),以及离子受体(IRs)、化学感受蛋白(CSPs)和气味结合蛋白(OBPs)的化学感受相关基因,均使用BLASTP(E<10−5)进行进一步注释。为了注释与味觉受体(GRs)和气味受体(ORs)相关的基因,研究人员通过TBLASTN鉴定了E值<10−5的候选基因座 ,并使用GeneWise预测基因结构(图4)。
表5 东方粘虫基因组功能注释统计
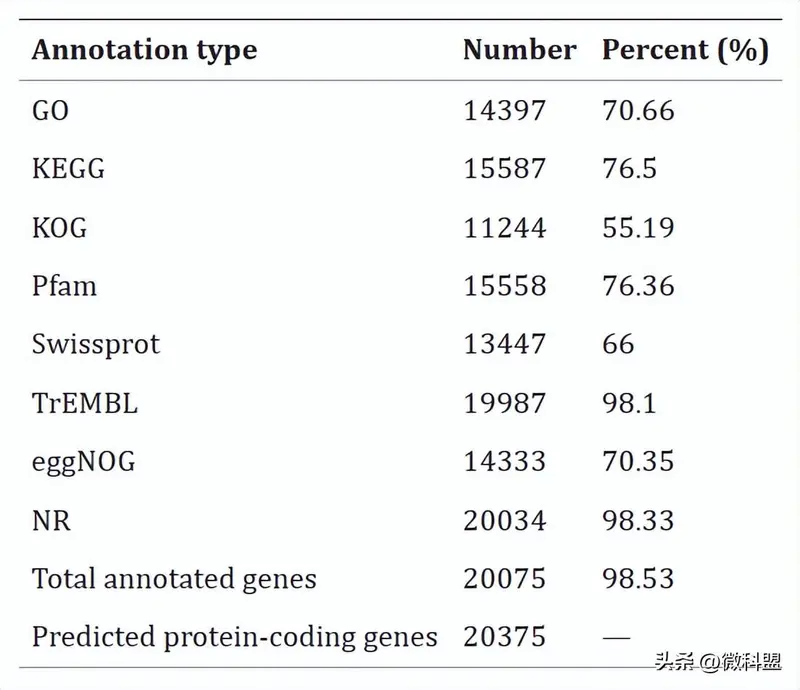
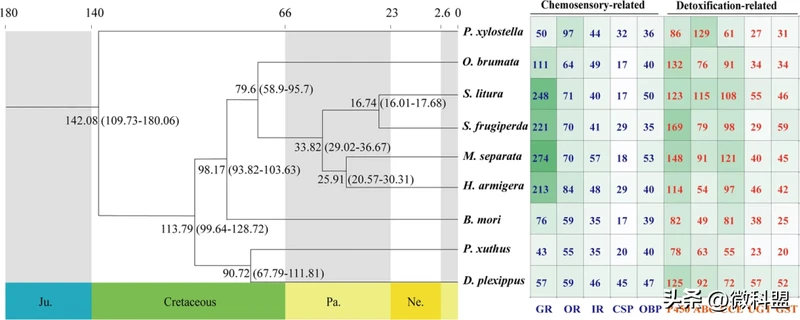
图4 东方粘虫和其他8种鳞翅目昆虫解毒和化学感觉基因的分化时间与分布。分支节点值表示物种间推断的分化时间。右侧小格中的数字表示每个物种中相应基因家族的规模。小格的底色越深,对应物种中编码的基因就越多。
系统发育分析
从NCBI下载了17种昆虫的蛋白质序列,用于系统发育分析(表6),其中包括8种鳞翅目(Lepidoptera)昆虫和9种与双翅目(Diptera)、鞘翅目(Coleoptera)、膜翅目(Hymenoptera)、半翅目(Hemiptera)和蜻蜓目(Ododata)相关的昆虫。使用OrthoFinder(v2.4.0)检测同源基因家族,并基于PANTHER数据库进行注释。使用MAFFT(v7.205)对单拷贝直系同源基因进行比对,并通过应用具有默认参数的Gblocks(v0.91b)去除模糊比对区域。系统发育树采用IQ-TREE(v1.6.10)构建,具有1000个bootstrap重复和LG+F+I+G4的**模型。根据从TimeTree数据库获得的化石记录,使用MCMCtree(PAML软件包)估计不同物种之间的分化时间(http://www.timetree.org/)。此外,采用CAFE(v5.0)软件,在p值阈值≤0.05的条件下,利用系统发育树结果(包括分化时间)鉴定基因家族的扩展和收缩。
表6 用于系统发育分析的17种昆虫的基因组下载链接
基因组共线性分析
为了对东方粘虫和草地贪夜蛾进行基因组共线性分析,在默认参数下使用Diamond(v0.9.29)鉴定了相似的基因对。通过MCScanX获得共线性区块中的所有基因,然后使用CIRCOS(v 0.69–9)在染色体上可视化共线性区块。东方粘虫和草地贪夜蛾之间只发现了一次裂变事件,这表明它们之间存在高度一致性(图5a)。
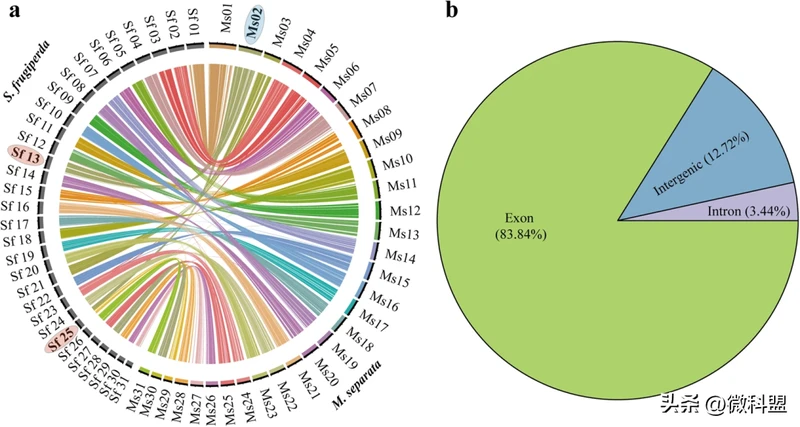
图5 东方粘虫基因组的基因组共线性和蛋白质编码基因的验证。(a)东方粘虫与草地贪夜蛾(Spodoptera frugiperda)的全基因组共线性分析。(b)RNA-seq纯净数据验证了蛋白质编码基因预测的准确性。
技术验证
基因组组装的评估
从三个方面验证基因组组装的完整性和准确性:首先,使用BWA将Illumina测序获得的纯净读长与基因组组装进行比对。结果显示,99.26%的Illumina读长与基因组组装一致。其次,核心真核基因定位方法(Core Eukaryotic Genes Mapping Approach,CEGMA)数据库包含458个保守的核心真核基因,其中431个(94.10%)在东方粘虫基因组中被鉴定出来。最后,采用BUSCO(v4)进行基因组组装完整性评估,参数为“-m prot -f -l eukaryota_odb9”,98.74%的保守核心BUSCOs存在于东方粘虫的基因组中。该结果表明研究人员获得了高质量的东方粘虫基因组组装。同时,本研究组装的contig N50为22.08 Mb,显著高于东方粘虫最近组装版本的7.31 Mb。研究人员组装的scaffold N50提高到23.00 Mb,略高于其它东方粘虫最近组装版本的22.68 Mb(表7)。
表7 5个东方粘虫基因组组装的比较统计
为了评估染色体组装的质量,将组装体剪切成100 kb的小箱(bins),并利用相互作用对的强度绘制热图。Hi-C热图显示,31条不同染色体沿对角线的相互作用强度明显高于非对角线位置的相互作用强度。
基因预测的评价
BUSCO分析也用于评估基因预测的结果。从基因组预测基因集中鉴定出了98.74%(942/954)的BUSCOs,略高于其它近期东方粘虫组装版本的98%和98.2%。同时,83.84%的RNA-seq数据与预测的外显子对齐(图5b)。这些结果证实了东方粘虫基因组基因预测的完整性和准确性。此外,在此次基因组组装中鉴定了20,375个蛋白质编码基因,显著多于最近可用的**参考基因组组装版本中的17,549个蛋白质编码基因。研究人员进一步使用E值<10−5的局部BLASTN比较了两个基因组组装中的蛋白质编码基因集。在两个基因组组装中共鉴定出16,398个蛋白质编码基因,2828个蛋白质编码基因仅在此次基因组组装中被鉴定。
比较基因组分析
18种昆虫共鉴定出27,002个同源基因家族,其中565个单拷贝直系同源基因家族被用于系统发育分析。系统发育分析结果表明,鳞翅目昆虫从其共同祖先进化而来的时间晚于双翅目、鞘翅目、膜翅目和半翅目(图6)。在鳞翅目中,东方粘虫和棉铃虫聚为一个分支,并在大约2529(2057-3031)万年前分化。此外,东方粘虫和草地贪夜蛾在大约3382(2902-3667)万年前发生分化。同时,GO富集分析显示,在东方粘虫基因组中的594个扩张基因家族主要涉及“DNA整合”(GO:0015074)、“核小体”(GO: 0000786)和“RNA依赖的DNA聚合酶活性”(GO:0003964),而1329个收缩基因家族主要涉及“信号转导调控”(GO:0009966)、“膜”(GO:0016020)和“丝氨酸型肽链内切酶活性”(GO:0004252)(图7,8)。
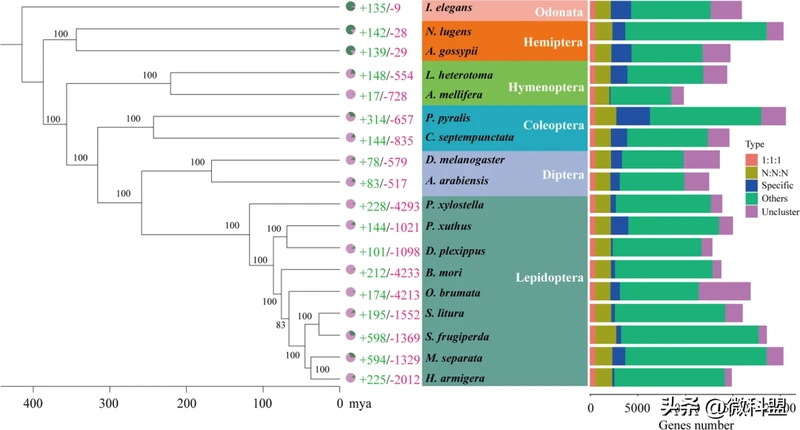
图6 东方粘虫与其它17种昆虫的系统发育树。基于565个单拷贝基因计算最大似然系统发育树。每个物种分支的右侧显示了扩张基因家族(绿色)和收缩基因家族(红色)的数量。彩色直方图表明,每个物种的基因可分为5组:1∶1:1(常见基因家族的单拷贝直系同源基因);N: N: N(相同基因家族中同一基因的多拷贝直系同源基因);特异性(来自每个物种的特异基因家族的基因);其他(不属于以上任何直系同源分类的基因);非聚类(未聚类到任何家族的基因)。
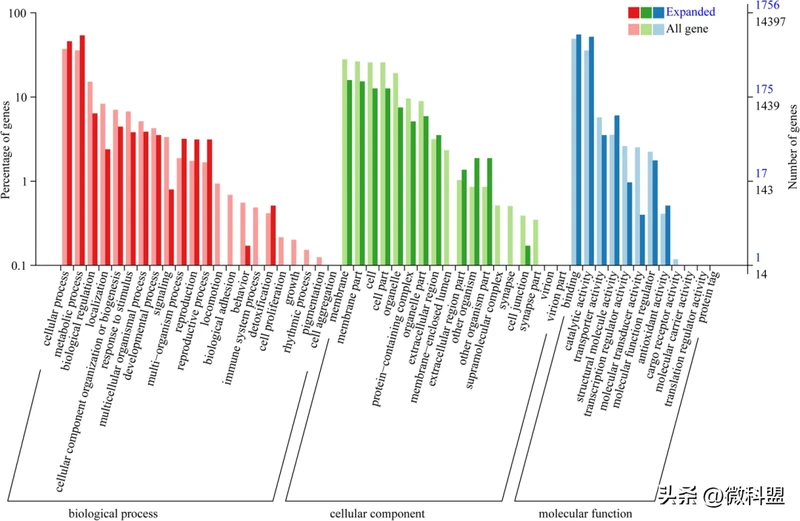
图7 东方粘虫扩张基因家族的Go富集分析
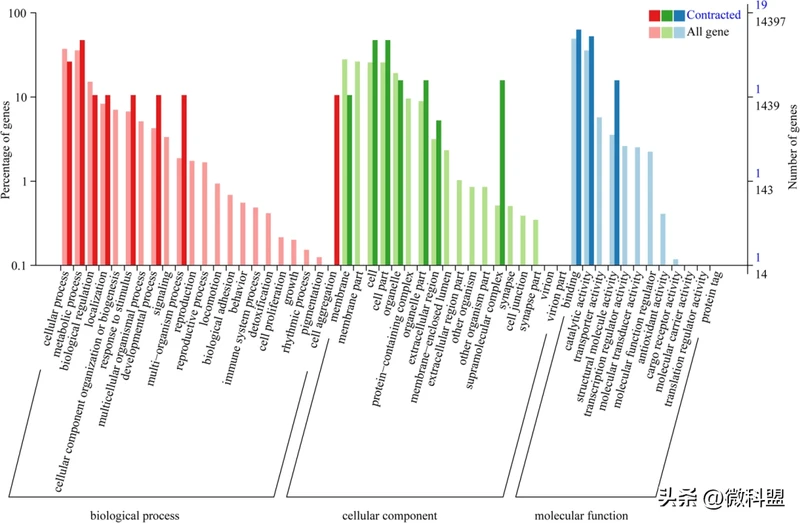
图8 东方粘虫收缩基因家族的Go富集分析